Will AlphaFold change bioscience research?
Above: A range of glycolytic enzymes visualised by David Goodsell, courtesy of PDB-101.
Google-backed AI platform AlphaFold has predicted 3D protein structures from DNA sequences with such accuracy that some regard the protein folding problem as ‘solved’. But what do these results mean for everyday life science research – and are protein crystallographers now out of a job?
December 4th 2020
The headlines this week have been of that rare excitable type not seen since the first news broke on CRISPR-Cas9 gene-editing. “This will change everything” declared Nature above a report on AlphaFold’s performance at a recent protein-structure prediction challenge. The MIT Technology Review declared that the AI programme has “solved” a 50-year-old grand challenge of biology.
So, is that it? Can anyone working in biology now simply type in a DNA sequence and visualise the resulting protein structures?
Not quite.
The news is based on AlphaFold’s very good results at a protein-structure prediction challenge known as CASP and a press release from DeepMind Technologies, AlphaFold’s UK-based and Google-owned developers. The release claims the unrivalled accuracy of AlphaFold will “dramatically accelerate progress in some of the most fundamental fields that explain and shape our world”, including drug discovery.
Structural biologists are excited about this clear step forward in protein-structure prediction, but have urged caution against hyperbole. Firstly, there are some blind spots in AlphaFold’s ability to visualise proteins in 3D: because its algorithms feed from the world’s databases of solved structures, it will not be able to predict proteins with folds that are not well represented in these data stores.
Also, the CASP tests did not include predictions of notoriously tricky “multi-protein” complexes and larger cellular structures like ribosomes. As these are some of the most interesting structures in biology, those who specialise in existing techniques to understand them experimentally are not likely to be out of a job any time soon.
AlphaFold’s accuracy at the latest CASP was indeed streaks ahead of other protein-modelling competition (see Fig.1) – predicting the structure of dozens of proteins with a margin of error of just 1.6 angstroms (Å), about the size of a chemical bond. But this still may not be quite accurate enough to make predictions on the atomic interactions required for drug discovery, said Stephen Curry, professor of structural biology at Imperial.
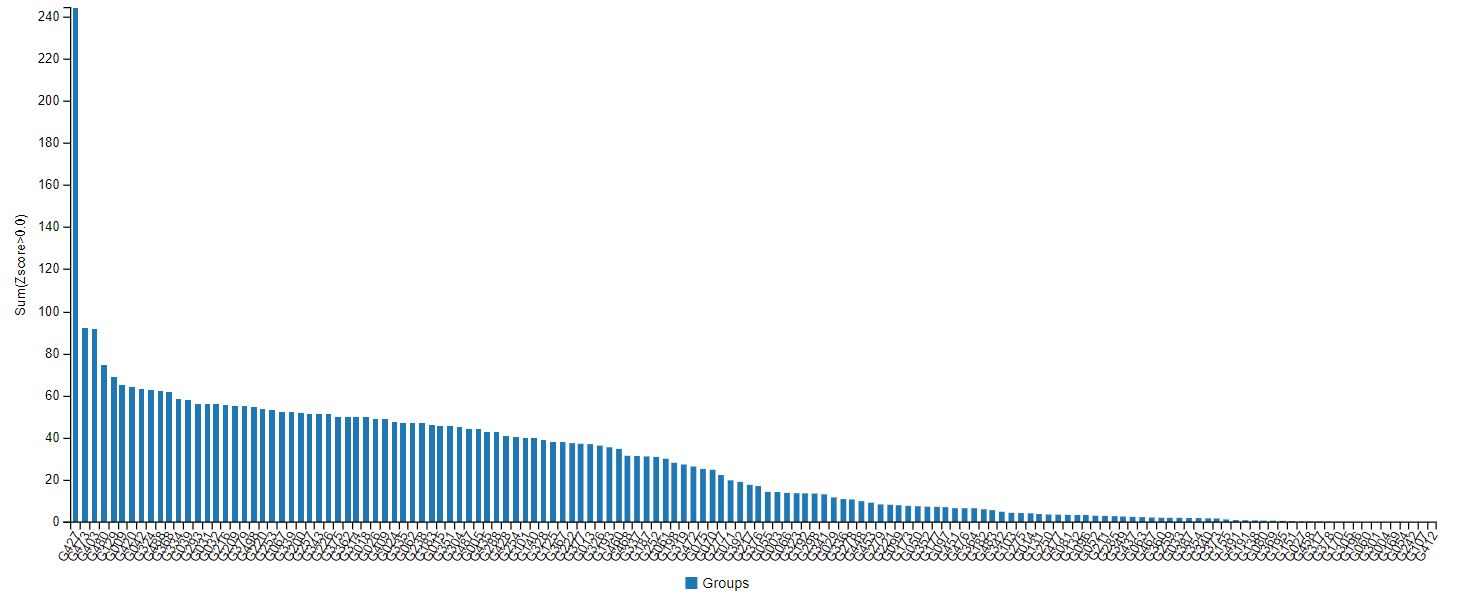 Fig.1: Results of the CASP14 protein-prediction challenge. G427, on the far left, is DeepMind’s Alphafold group.
Fig.1: Results of the CASP14 protein-prediction challenge. G427, on the far left, is DeepMind’s Alphafold group. “It shows the power of the method to predict the overall fold,” said Curry. “But I don’t think it includes the side-chain atoms, which are often key for drug interactions. Therefore, the accuracy figure for all atoms (backbone and side-chain) is likely to be much greater than 1.6Å. For drug discovery you probably want to be confident of atomic positions within a margin of less than 0.3Å. We’re not there yet.”
Peijun Zhang, professor of structural biology and Welcome Trust investigator at Oxford’s Nuffield Department of Medicine, said she was very excited at the results, which represented a “gigantic leap” forward since the last CASP challenge in 2018.
But she said if she had access to the tool she would still validate its predicted structures using experimental techniques.
“I would like to use it, definitely. But like most computational results, we would still like to validate it in a biological context.”
Zhang predicted that AlphaFold would be a very useful complementary tool to ease some of the more “tedious” steps in understanding protein structures of large macromolecular complexes and machines in cellular contexts, a problem that still requires experimental approaches, in particular cryo-EM and electron tomography.
“A lot of structural biologists might be thinking that they might be out of a job soon! I don’t think we are anywhere close to this. Structures like ribosomes and photosynthesis centres are huge and complex in comparison. How the many different parts fit together to form a functional machine is still a big challenge for AI in the near future.”
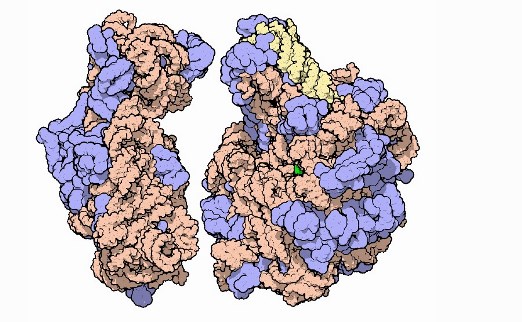 Ribosomal subunits, with RNA in orange and yellow and proteins in blue. It's unclear if AlphaFold is able to predict the structure of multi-protein complexes or macromolecular 'machines' made up of several protein and non-protein subunits. Image courtesy of David Goodsell and PDB-101.
Ribosomal subunits, with RNA in orange and yellow and proteins in blue. It's unclear if AlphaFold is able to predict the structure of multi-protein complexes or macromolecular 'machines' made up of several protein and non-protein subunits. Image courtesy of David Goodsell and PDB-101.Paul Freemont, chair in protein crystallography and head of the Section of Structural and Synthetic Biology at Imperial, said that “although there are several large caveats”, the sheer leap in performance of AlphaFold from other protein prediction groups does represent a “landmark moment” for bioscience.
“For a significant number of proteins, the use of algorithms like AlphaFold to determine their three-dimensional structure will likely become the norm and these structural models will provide the biochemical details for many new biological systems.”
He agreed that initially, AlphaFold will join a suite of other physical and computational tools used to determine macromolecule and cellular structures at different scales, combining with techniques like cryo-tomography to create near atomic resolution maps of parts of cells.
“Protein crystallography will still be used for determining more complex multi-protein complexes, and in drug screening to experimentally verify any predictions through regulatory needs,” said Freemont. “Although this may change.”
Predicting 3D structures is not the only thing biologists need to know about proteins.
“The next big question will be to predict how proteins move and function – they are not static entities,” said Sheena Radford OBE, Director of the Astbury Centre for Structural Molecular Biology and the University of Leeds.
“Plus, predicting why mutations change folding and function – and hence how disease is caused – are also important next steps. This work tells us about the final structure, but not how the structure was found from the billions of possibilities – that is really the protein folding problem. Perhaps AI can also solve that in due course.”
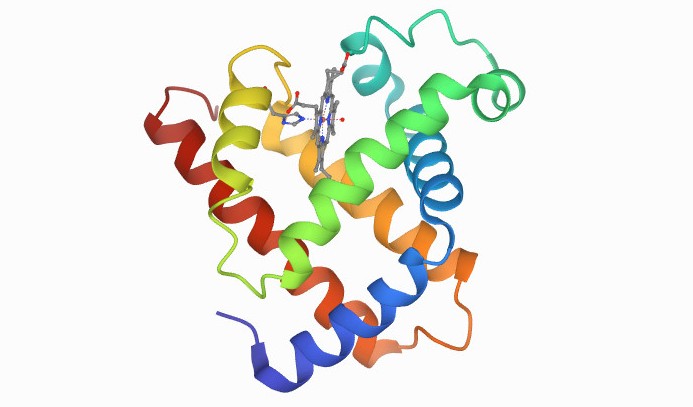 The first protein to have its atomic structure and 3D shape determined was myoglobin (pictured), in the late 1950s. Calculating the structure required many years of x-ray crystallography work by John C Kendrew, who won the Nobel Prize for his achievement in 1962. Google's AlphaFold would be able to deduce this structure from its corresponding DNA sequence in minutes, if not seconds. Image courtesy of PBD-101.
The first protein to have its atomic structure and 3D shape determined was myoglobin (pictured), in the late 1950s. Calculating the structure required many years of x-ray crystallography work by John C Kendrew, who won the Nobel Prize for his achievement in 1962. Google's AlphaFold would be able to deduce this structure from its corresponding DNA sequence in minutes, if not seconds. Image courtesy of PBD-101. There are around 200,000 known or 'solved' protein structures, but 180 million known protein sequences, said Professor Jackie Hunter, a director of drug discovery company BenevolantAI and former CEO of the Biotechnology and Biological Sciences Research Council..
She believes AlphaFold may help us better understand the structure of proteins that have been difficult to determine experimentally, such as membrane-bound proteins, and the time and labour saved by its insights as it improves will help advance many areas of bioscience.
“This will allow bioscientists to make progress across in a wide range of areas from microbiology through synthetic biology to a better understanding of diseases.”
More information and data on AlphaFold and its performance can be found here.
Tom Ireland MRSB is editor of The Biologist and head of publications at the Royal Society of Biology.
